Adrenogenitales Syndrom (AGS)
12.01.18 - Wechsel von NaCl 0.9% --> Ringerfundin/Ringeracetat als Infusionslösung
- Grundlagen
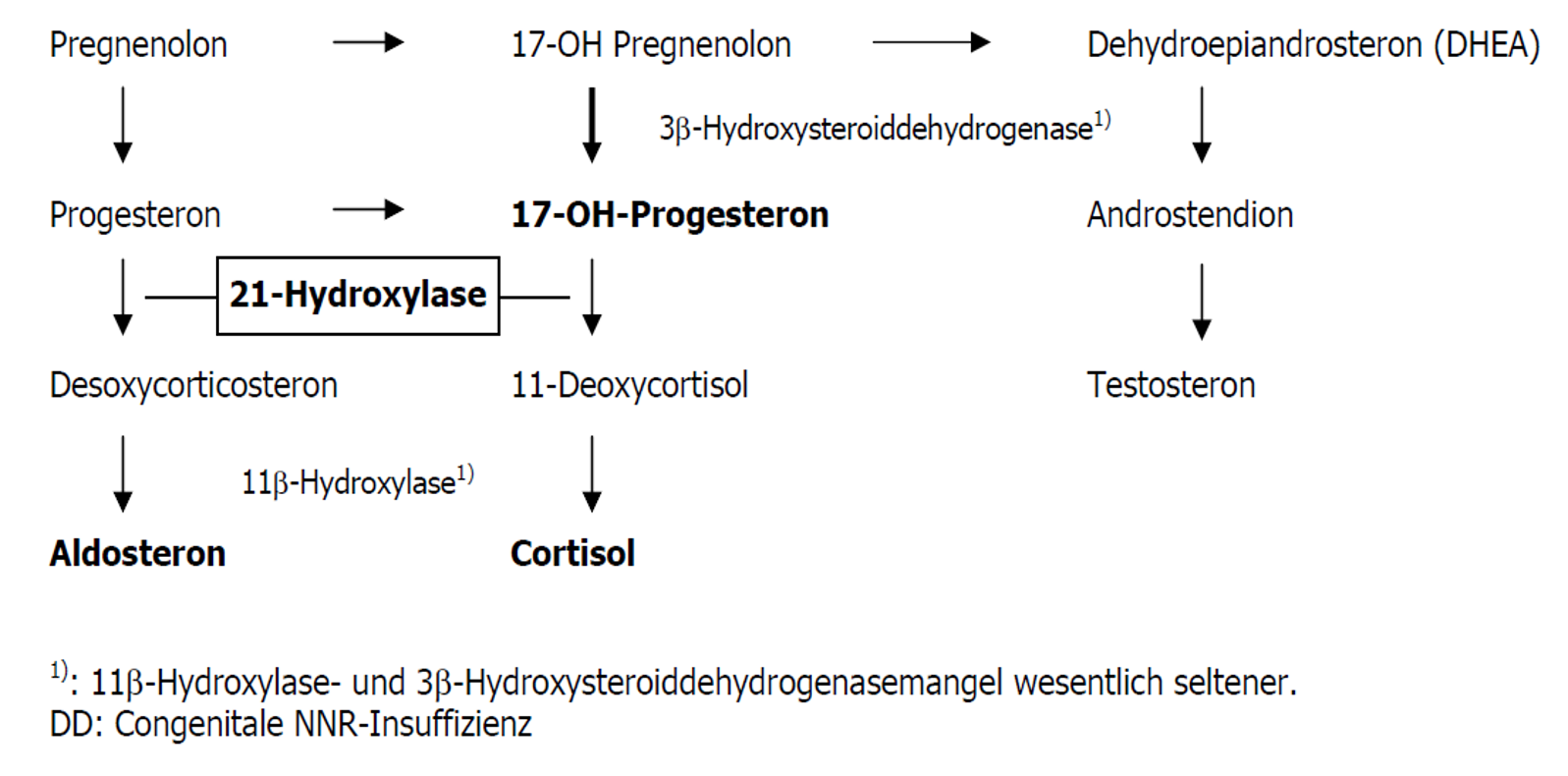
- Abbildung 1: Vereinfachte Darstellung der Steroidbiosynthese
- Screening
- Klinische Symptome
- Diagnostik (V.a. AGS ist ein Notfall)
- Therapie
- Verlaufskontrollen
- Abbildung 2: Algorithmus für AGS Screening bei Neugeborenen (NEJM August 2003)
Autoren: S. El Helou / D. l'Allemand
Version: 11/07, Rev. P. Tonella 09/13
Grundlagen
Der Terminus AGS beschreibt mehrere autosomal rezessiv vererbte Störungen, welche eine teilweise oder komplette Defizienz der Enzyme aufweisen, die an der Synthese von Cortisol und/oder Aldosteron beteiligt sind. In 95% der Fälle handelt es sich um ein AGS bei 21-Hydroxylase Mangel, bei 75% davon besteht ein Salzverlustsyndrom. Inzidenz: 1:15000 in der Schweiz.
Abbildung 1: Vereinfachte Darstellung der Steroidbiosynthese
Screening
AGS Screening beim Neugeborenen im Rahmen des Stoffwechselscreenings auf Filterpapierkarte (siehe Anhang):
17-OH Progesteron ↑↑ (Norm für TG im Labor (derzeit Zürich): < 30 nmol/L, FG: Gestationsalterabhängig).
Klinische Symptome
Abhängig von Art und Schwierigkeit der Enzymdefizienz, Geno-/Phänotyp Korrelation:
- Virilisierung Knaben: Genitale normal bis gross, evtl hyperpigmentiert. Prämature Adrenarche
- Virilisierung Mädchen: Klitorishypertrophie ohne/mit Sinus urogenitalis bis fast vollständige Virilisierung ("peno-/scrotale Hypospadie" mit "Kryptorchismus", Prader Stadium I-IV; Pseudo-Pubertas praecox, Prämature Adrenarche
- Salzverlust: Am Ende der 1. Lebenswoche Hyponatriämie, Hyperkaliämie, Exsikkose, hypovolämischer Schock, Erbrechen
- Glukokortikoidmangelzeichen (seltener): Hirnödem, Blutdruckabfall, Hypoglykämie
- Addison-Krise bei Untersubstitution und Krankheit, setzt sich aus vorgenannen Zeichen zusammen
- Wachstumsbeschleunigung und zunehmende Virilisierung bei Untersubstitution
Diagnostik (V.a. AGS ist ein Notfall)
Bei Salzverlut / Addison Krise:
- Serum/Plasma: Na, K, Cl, BGA, BZ und per A-Post nach Basel: 17-OH Progesteron, Androstendion, Testosteron, Renin, evtl. 11-beta-Desoxycortisol, DHEA
- Urin: Steroidprofil (KISPI Zürich), Na, K, Kreatinin
Ultraschall: Nebennieren-Hyperplasie?
Endokrinologisches Konsil (eilig!)
Zusätzlich bei Störung der Sexualdifferenzierung:
- Ultraschall: Uterus/inneres Genitale?
- Karyotyp (Frage nach Y-Chromosom per FISH-Analyse)
- Kinderchirurgisches Konsilium
- evtl. Einschaltung KJPD
Therapie
Glucocorticoid: Hydrocortison (Solu-Cortef®); Hydrocortisone p.o. a 10 mg (evtl. Zubereitung 1 und 2 mg Kapseln)
Mineralocorticoid: Fludrocortison a 0.1 mg (Florinef®); Aldosteron i.v.
Akute Notfallsituation (meist Addison-Krise)
- Bei arterieller Hypotension Ringerfundin, 20 ml/kgKG rasch i.v. (max. 1h)
- Dauerinfusion mit Ringeracetat mit 1 oder 5% Glucose, je nach Natriumsubstitutionsbedarf (CAVE Hypoglykämie, Natriumausgleich zu schnell (Anstieg max. 0.5-1 mmol/l/h)
- EKG-Monitoring bei Hyperkaliämie
- Hydrocortison: Ladedosis 100 mg/m2 i.v./i.m. (äquivalent: 20 mg/m2 Methylprednisolon oder 2 mg/m2 Dexamethason*)
*bei unklarer Diagnose am besten Dexamethason, interferiert nicht mit biochemischer Diagnostik (CAVE Dexamethason keine mineralocorticoide Wirkung)
- Fludrocortison: 0.1-0.2 mg/Tag p.o. bei guter GI Funktion (CAVE Dosierung unabhängig vom Körpergewicht)
Erhaltungstherapie
- Hydrocortison: 10-15 mg/m2 /Tag in 3 Einzeldosen (im Säuglingsalter als 1-2 mg Kapseln, nicht in Spritze auflösen)
- Fludrocortison: 0.05-0.2 mg/Tag p.o. (CAVE Hypertonus, Dosierung unabhängig von Körpergewicht), aufgeteilt in 2 Dosen
- Im ersten Lebensjahr ist zusätzlich zum Fludrocortison NaCl p.o. (1-2 g/Tag (17-24 mmol/Tag) auf mehrere Mahlzeiten aufgeteilt) notwendig
Bei der klinischen Einstellung wird eine Balance gesucht zwischen Hyperandrogenismus (bei Untersubstitution) und Hypercortisolismus (bei Übersubstitution)
Stressdosis (Operation, febriler Infarkt)
- Bei Fieber zwischen 38°C und 39°C: Hydrocortison-Erhaltungstherapie verdoppeln
- Bei Fieber über 39°C oder Operation: Hydrocortison-Erhaltungstherapie verdreifachen
- Fludrocortison bleibt bei allen Stress-Situationen gleich
- Bei i.v.-Verabreichung: Säuglinge und Kleinkinder Solu-Cortef 25 mg i.v. 6-stündlich, Schulkinder Solu-Cortef 50 mg i.v. 6-stündlich oder gemäss unten stehendem Schema
| Hydrocortison (Solu-Cortef®) | ||
| Alter | Bolus | Erhaltung/24 Stunden |
| ≤ 3 Jahre | 25 mg i.v. | 25-30 mg i.v. |
| > 3 aber < 12 Jahre | 50 mg i.v. | 50-60 mg i.v. |
| ≥ 12 Jahre | 100 mg i.v. | 100 mg i.v. |
Bolus + Erhaltung entspricht 100 mg/m2; nach Harriet Lane 18th Edition (2009) S 1031 kann diese Dosis auch als Dauertropf verabreicht werden.
Referenz: Nils P. Krone, Birmingham. MTE Session, ESPE 2013
Verlaufskontrollen
- Elektrolytkontrollen: initial individualisiert (z.B. 1-2 wöchentlich) (CAVE Salzverlustsyndrom kann sich erst spät demaskieren (nach 1-4 Wochen oder auch erst später im Rahmen einer Erkrankung))
- Therapiesteuerung (individualisiert z.B. alle 3 Monate)
17-OH Progesteron (Ziel: Teilsuppression ≤ 30 nmol/l)
Androstendion, Testosteron (Ziel: alters- und geschlechtsspez. Normbereich)
Gewicht, Länge, Wachstumsgeschwindikeit
Blutdruck
Pubertätsstadium nach Tanner
Knochenalter
Abbildung 2: Algorithmus für AGS Screening bei Neugeborenen (NEJM August 2003)
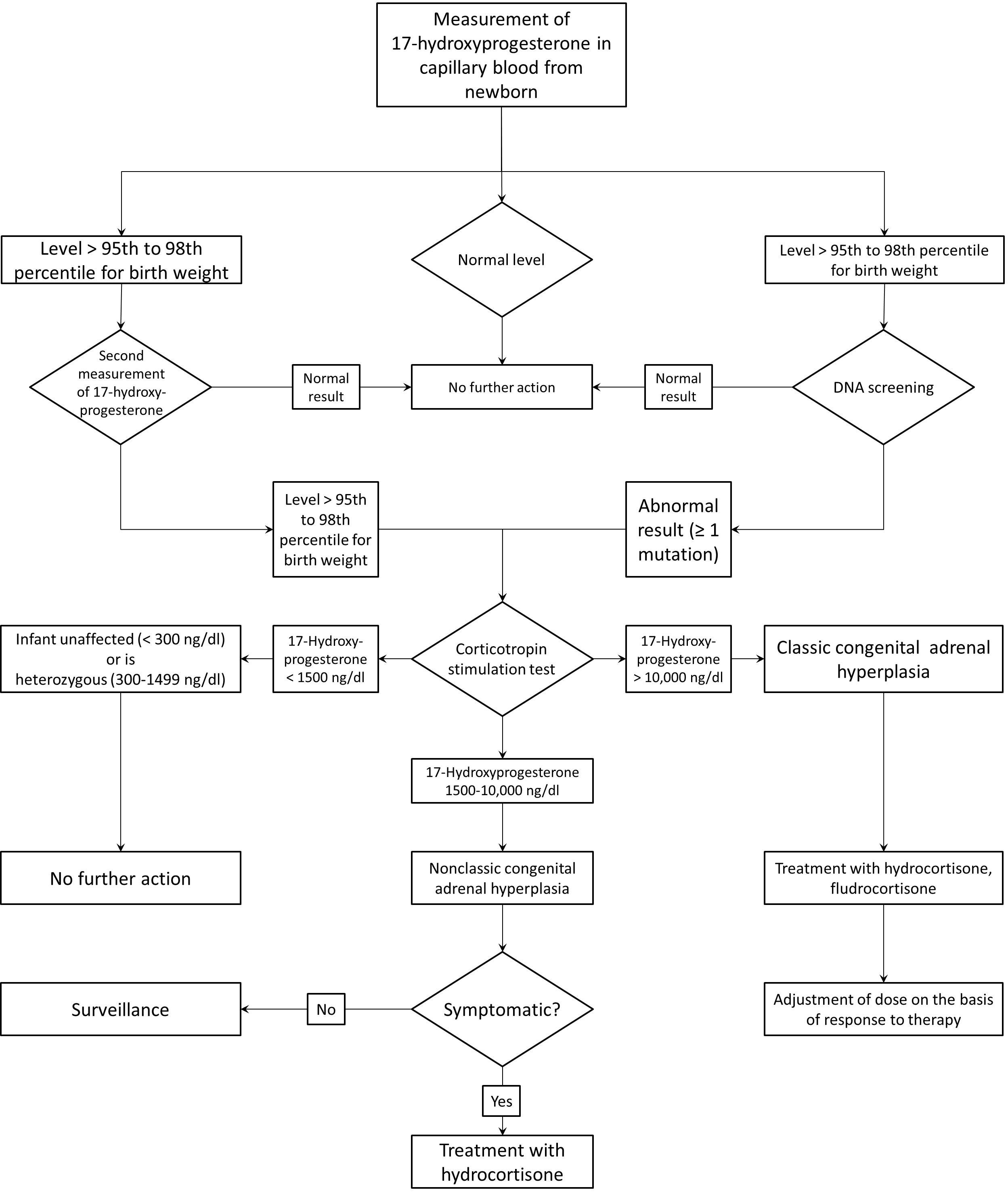
Protocols vary from center to center. Screening is first performed on dried capillary blood. A 17-hydrxyprogesterone level in the 95th to 98th percentile for birth weight prompts a second screening test, which may be either a repeated hormonal assay or a molecular test to determine whether the infant is a carrier of at least one common mutation in the gene for 21-hydroxylase (CYP21). If the results of either of these tests are abnormal, the infant undergoes a corticotropin (cosyntropin) stimulation test. Depending on results, the infant is classified as unaffected, a probable heterozygote, having nonclassic disease, or having classic disease. The need for further management is dictated by these results.
Referenzen
- Joint ESPE/LSWPES CAH Working Group. Consensus Statement on 21-Hydroxylase Deficiency; Horm Res 2002; 58; 188-195
- Speiser PW, White PC. Congenital Adrenal Hyperplasia. NEJM 2003; 349: 776-788
- Wilson TA. Congenital Adrenal Hyperplasia. eMedicine 2004
- Speiser PW et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2010;95:4133-4160 (Abstract)